再生医療製品の製造において、無菌性の確保は患者様の生命に直結する最重要課題です。しかし、どれほど厳格に管理された細胞培養加工施設(CPC)であっても、目に見えない微粒子や微生物による汚染リスクをゼロにすることは容易ではありません。特に、最終製品に対する無菌試験だけでは、製品全体の無菌性を完全には保証できないというジレンマがあります。そこで不可欠となるのが、科学的根拠に基づいた「無菌操作の基本原則」の徹底と、精緻な「リスク評価」の実践です。
本記事では、GCTP省令や関連ガイドラインを踏まえ、現場の製造管理者や品質保証担当者の方々に向けて、無菌管理体制を強固にするための具体的なアプローチを解説いたします。リスクベースアプローチによる管理手法を再確認し、より安全で信頼性の高い製造プロセスを構築するための一助となれば幸いです。
再生医療における無菌操作の基本原則とリスク評価の結論

再生医療等製品の製造において、無菌性を担保するために最も重要なのは、最終的な検査結果だけに頼るのではなく、プロセス全体で品質を作り込むという考え方です。ここでは、現代の無菌操作管理において中心的な概念となるリスクベースアプローチと、その背景にある法的・科学的根拠について解説いたします。
無菌性保証における「リスクベースアプローチ」の重要性
無菌性保証において、従来の「経験と勘」に頼る管理から脱却し、科学的知見に基づいた「リスクベースアプローチ」を採用することが世界的な標準となっています。これは、製造工程に潜む汚染リスク(ハザード)を特定し、その発生確率や影響度を評価した上で、リスクの程度に応じた適切な管理策を講じる手法です。
すべての工程を均一に管理するのではなく、汚染リスクが高い重要な工程(クリティカルプロセス)にリソースを集中させることで、効率的かつ効果的に無菌性を担保できます。ICH Q9(品質リスクマネジメント)のガイドラインに沿って、主観を排除した客観的な評価を行うことが、規制当局への説明責任を果たす上でも極めて重要です。
最終製品での無菌試験の限界とプロセス管理の必然性
無菌操作において認識しなければならない重大な事実は、「最終製品の無菌試験に合格したからといって、そのロット全体の無菌性が完全に証明されたわけではない」という点です。無菌試験は破壊検査であり、また統計的なサンプリング検査に過ぎないため、検出感度には限界があります。
したがって、最終製品の試験結果はあくまで品質確認の一部に過ぎません。原材料の受入から製造、包装に至るまでの全プロセスにおいて、微生物混入のリスクを排除する「プロセス管理」こそが、無菌性保証(Sterility Assurance)の本質となります。プロセスバリデーションや日常のモニタリングデータが積み重なって初めて、製品の安全性を主張できるのです。
GCTP省令が求める構造設備と手順のハード・ソフト両面の整合性
GCTP省令(再生医療等製品の製造管理及び品質管理の基準に関する省令)では、無菌操作を行うためのハード(構造設備)とソフト(手順・管理)の両面が整合していることを求めています。高性能なアイソレーターや安全キャビネットを導入していても、それを運用する手順や作業者の行動が不適切であれば、無菌性は維持できません。
逆に、熟練した作業者がいても、空調システムや清浄度区分が不十分であれば汚染リスクは高まります。リスク評価においては、設備性能の限界と人的操作の不確実性の両方を考慮し、それらが相互に補完し合うようなシステムを構築する必要があります。ハードとソフトのバランスが取れた管理体制こそが、法令遵守と品質確保の鍵となります。
無菌操作を構成する4つの基本原則(4M)

無菌操作を成功させるためには、製造に関わる4つの要素(4M)を漏れなく管理することが不可欠です。ここでは、汚染リスクの主要因となり得るPersonnel(人)、Premises(設備)、Materials(原材料)、Methods(方法)のそれぞれについて、基本原則と管理のポイントを詳しく見ていきましょう。
Personnel(人):最大の汚染源である作業者の教育と行動管理
無菌操作エリアにおいて、最大の汚染源となり得るのは間違いなく「人(Personnel)」です。作業者の皮膚、毛髪、呼気からは常に微粒子や微生物が放出されており、動作によってその発塵量は大きく変動します。そのため、徹底した更衣手順(ガウニング)の習得と、無菌エリア内での緩やかで慎重な動作の徹底が求められます。
また、単に手順を教えるだけでなく、「なぜその動作が必要なのか」という微生物学的根拠を理解させる教育が重要です。体調不良時の申告制度や、定期的なガウニング評価を通じて、作業者自身の意識とスキルを高く維持することが、汚染リスク低減の第一歩となります。
Premises(設備):清浄度区分と気流制御による環境維持
「設備(Premises)」に関しては、作業内容に応じた適切な清浄度区分(グレードA〜D)の設定と維持が基本原則です。特に重要なのが、異なる清浄度区域間の差圧管理と気流制御です。清浄度の高い区域から低い区域へ空気が流れるように陽圧を保ち、汚染物質の流入を防ぐ必要があります。
また、HEPAフィルターを通した清浄空気による一方向流(ラミナーフロー)が、作業エリア(グレードA)で適切に維持されているかを定期的に検証することも欠かせません。スモークスタディによる気流可視化を行い、空気の淀みや乱流が生じていないかを確認することで、環境由来の汚染リスクを可視化し対策を講じることができます。
Materials(原材料):資材の滅菌保証と搬入時の除染プロセス
細胞培養に使用する培地、試薬、消耗品などの「原材料(Materials)」は、CPCへの搬入前に確実に無菌性が保証されていなければなりません。既滅菌製品を使用する場合は、その滅菌証明書(CoA)の確認に加え、包装の完全性をチェックすることが重要です。
さらに、外部から管理区域へ物品を持ち込む際の「除染プロセス」は、汚染持ち込みの重大なリスクポイントです。パスボックスでの清拭消毒や気化過酸化水素(VHP)などによる除染が、バリデーションされた手順通りに行われているか、また除染剤の残留が製品に影響を与えないかどうかも、厳密に管理する必要があります。
Methods(方法):バリデーションされた標準作業手順書(SOP)の遵守
適切な「方法(Methods)」とは、科学的に妥当性が検証(バリデーション)され、文書化された標準作業手順書(SOP)を指します。無菌操作は属人化を防ぐため、誰が行っても同じ品質が保てるよう、手順が具体的かつ明確でなければなりません。
SOPには、単なる操作手順だけでなく、異常時の対応や使用する器具の配置、消毒の頻度なども詳細に規定する必要があります。そして最も重要なのは、そのSOPが現場で厳守されていることです。手順からの逸脱は直ちに汚染リスクに直結するため、逸脱管理システムを通じて再発防止策を講じ、SOP自体も定期的に見直しを行う継続的な改善サイクルが求められます。
細胞培養加工施設(CPC)におけるリスク評価の実施フロー

リスク評価は一度実施して終わりではなく、製品ライフサイクル全体を通じて継続的に行うべき活動です。ここでは、細胞培養加工施設(CPC)において具体的にどのようにリスクマネジメントを進めるべきか、その標準的なフローを5つのステップに分けて解説いたします。
リスク特定:プロセスごとのハザード(汚染源)の洗い出し
リスクマネジメントの最初のステップは、「何がうまくいかない可能性があるか」を見つけ出すリスク特定です。製造プロセスのフローチャートを基に、各工程における潜在的なハザード(危害の源)を洗い出します。
例えば、「培地交換時のピペット操作での接触」「インキュベーターのドア開閉による温度変化」「原材料搬入時の外装汚染」など、具体的かつ網羅的にリストアップすることが重要です。この段階では、過去の逸脱事例やヒヤリハット報告、現場作業者の経験知などを積極的に活用し、些細と思われるリスクも見逃さないようにしましょう。
リスク分析:FMEA法などを用いた発生確率と重大性の数値化
特定されたリスクに対し、そのリスクが顕在化した場合の影響度を分析します。一般的にはFMEA(故障モード影響解析)などの手法を用い、「発生確率(Probability)」「重大性(Severity)」「検出可能性(Detectability)」の3つの指標でスコアリングを行います。
- 発生確率: その失敗がどの程度の頻度で起こり得るか
- 重大性: 汚染が発生した場合、患者や製品にどの程度の影響を与えるか
- 検出可能性: 汚染やミスが発生した際、現在の管理体制でそれを発見できるか
これらを数値化し、リスク優先度数(RPN)を算出することで、客観的なリスクの大きさを把握できます。
リスク評価:許容範囲の決定と対策優先順位の策定
算出されたリスクスコアに基づき、そのリスクが「受容可能か」あるいは「低減措置が必要か」を判断します。あらかじめ組織としてリスクの受容基準(許容範囲)を定めておくことが必要です。
例えば、RPNが一定値を超える項目や、重大性が「高」と判定された項目については、優先的に対策を講じる必要があります。すべてのリスクをゼロにすることは現実的ではないため、限られたリソースの中でどのリスクから対処すべきか、優先順位を明確に決定するプロセスがこの評価段階となります。
リスクコントロール:エンジニアリング対策と管理的対策の適用
受容できないリスクに対して、具体的な低減策を実行します。対策には大きく分けて「エンジニアリング対策(設備的対策)」と「管理的対策(ソフト的対策)」があります。
エンジニアリング対策としては、閉鎖系システム(クローズドシステム)の導入やアイソレーターの使用などが挙げられ、これらは人為的ミスを物理的に防ぐ効果が高いです。一方、管理的対策には、SOPの改訂、ダブルチェックの導入、教育訓練の強化などが含まれます。可能な限り、人の注意のみに頼らない設備的な対策を優先することが、堅牢なリスクコントロールにつながります。
リスクレビュー:定期的な再評価と変更管理時の見直し
リスク対策を実施した後も、その効果を定期的に確認し、新たなリスクが生じていないかをレビューし続ける必要があります。これをリスクレビューと呼びます。特に、製造設備や手順の変更(変更管理)、新たな知見が得られた場合、あるいは年次レビューのタイミングなどで実施します。
リスクマネジメントは静的な文書ではなく、生きたプロセスです。モニタリングデータや逸脱の傾向分析(トレンド分析)の結果をフィードバックし、リスク評価書を常に最新の状態に保つことで、継続的な品質改善(Continual Improvement)を実現しましょう。
具体的な汚染リスク要因と低減対策の事例

理論的なリスク評価手法を理解したところで、実際のCPC現場で頻繁に課題となる具体的な汚染リスク要因と、それに対する実践的な低減対策を見ていきましょう。現場の状況と照らし合わせながら確認してみてください。
安全キャビネット内での気流妨害による交差汚染リスク
安全キャビネット(BSC)は無菌操作の要ですが、作業者の腕や物品の配置によってエアカーテンが乱れ、外気が巻き込まれるリスクがあります。特に、吸込口(フロントグリル)を腕や物で塞いでしまうと、バリア機能が著しく低下し、交差汚染の原因となります。
対策:
- キャビネット内には必要最小限の物品のみを持ち込む
- 吸込口の上に物を置かないよう徹底する
- 急激な手の出し入れを避ける
- スモークスタディで気流の乱れがない配置を検証・固定化する
これらをSOPに明記し、実地訓練で気流の特性を体感させることが有効です。
パスボックスを介した物品搬入出時の除染不全リスク
パスボックスは管理区域内外をつなぐ接点であり、ここでの除染が不十分だと汚染物質がグレードの高い区域へ侵入してしまいます。特に、物品の裏側や重ねた部分への消毒液の行き渡り不足、接触時間(ウェッティングタイム)の不足がよくあるリスクです。
対策:
- 物品を詰め込みすぎず、消毒剤が全面に接触するように配置する
- スプレー噴霧だけでなく、清拭による物理的な除去を併用する
- 消毒剤の接触時間をタイマーで管理する手順を導入する
- 可能であれば、UV殺菌灯だけでなくVHP等のより確実な除染方法を検討する
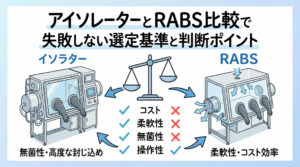
アセプティックコネクター使用時と開放系操作の比較評価
チューブ接続において、従来の開放系操作(火炎滅菌下での接続など)は、作業者の技量に依存する部分が大きく、環境由来の汚染リスクも高くなります。これに対し、無菌接合装置やアセプティックコネクターの使用はリスクを大幅に低減できます。
比較評価:
| 項目 | 開放系操作 | アセプティックコネクター |
|---|---|---|
| 環境依存度 | 高い(グレードA必須) | 低い(グレードC/Dでも可の場合あり) |
| スキル依存度 | 高い(熟練が必要) | 低い(標準化しやすい) |
| 汚染リスク | 相対的に高い | 非常に低い |
リスク評価の結果、開放系操作のリスクが許容できない場合は、シングルユース製品やコネクター技術を活用した閉鎖系への移行を強く推奨します。
グレードA/Bエリアでの作業者の発塵とガウニングの不備
グレードA/Bエリアであっても、作業者が存在する限り発塵は避けられません。特に、手首部分や顔周りの隙間からの発塵、あるいは不適切なサイズの無菌衣着用による動作時のポンピング現象(衣服内の空気が押し出される現象)がリスクとなります。
対策:
- 無菌衣の着用前に鏡で隙間がないか確認する手順(バディチェック)の導入
- 手首部分は滅菌テープで確実にシールする
- 作業者の体格に合ったサイズの無菌衣を選定する
- 更衣バリデーションを定期的に実施し、個人の癖を修正する
これらを徹底し、環境モニタリングデータと紐づけて作業者の評価を行うことが重要です。
インキュベーターの開閉に伴う環境変化と汚染リスク
CO2インキュベーターは細胞にとって最適な環境ですが、同時にカビや細菌にとっても増殖しやすい環境です。ドアの開閉による庫内温度・湿度の変化で結露が生じると、そこがカビの温床となるリスクがあります。また、トレイの出し入れ時に手袋が庫内壁面に触れることによる接触汚染も懸念されます。
対策:
- ドアの開閉回数と開放時間を最小限にする
- 庫内の清掃・消毒スケジュールを厳格化する
- 加湿水(使用する場合)は滅菌水を使用し、頻繁に交換する
- 複数の患者検体を扱う場合は、インキュベーターを分けるか、密閉容器を使用する
日常的な庫内観察と、定期的な除染サイクルの確立が不可欠です。
リスク評価に基づくプロセスシミュレーション(培地充填試験)

リスク評価の結果が妥当であるか、そして構築された無菌操作手順が実際に機能しているかを検証する最終的な手段が「プロセスシミュレーション(培地充填試験)」です。これは、実際の製品の代わりに微生物が増殖する培地を用いて製造工程を模倣する試験で、無菌操作の適格性を証明するために必須のプロセスです。
最悪条件(ワーストケース)の設定根拠
プロセスシミュレーションでは、日常の製造条件だけでなく、無菌性にとって最もリスクが高い条件、すなわち「ワーストケース」を含めて実施する必要があります。これは、最悪の条件下でも無菌性が保たれることを証明するためです。
ワーストケースの設定例:
- 最大作業時間: 許容される最長の作業時間で行う
- 最大人数: エリア内に滞在する最大の作業人数で行う
- 介入操作: 設備の調整や部材の補充など、日常起こり得る介入操作をあえて組み込む
- 疲労度: 作業シフトの後半や長時間作業後を想定する
リスク評価で特定された重要工程や変動要因を根拠として、これらの条件をプロトコルに設定します。
許容基準の設定と不合格時の原因究明プロセス
培地充填試験の許容基準は、原則として「汚染なし(陽性本数ゼロ)」です。万が一、培地に微生物の増殖(混濁)が認められた場合は、試験は不合格となり、直ちに原因究明プロセスを開始しなければなりません。
原因究明のステップ:
- 微生物の同定: 検出された菌種を特定し、環境モニタリング菌株や作業者の常在菌と比較する。
- プロセスの再調査: 録画データの確認や作業者へのヒアリングを行い、手順の逸脱や異常がなかったか確認する。
- リスク評価の見直し: 想定外のリスクがなかったか再評価する。
原因が特定され、是正措置(CAPA)が完了し、再試験で無菌性が証明されるまでは、実際の製品製造を再開することはできません。
定期的な再バリデーションの頻度とタイミング
プロセスシミュレーションは、新規立ち上げ時の初期バリデーション(通常3ロット連続成功)だけでなく、定期的な再バリデーション(Re-validation)が必要です。一般的には、半年(6ヶ月)に1回程度の頻度で実施することが推奨されています。
また、以下のような変更があった場合にも、臨時の再バリデーションが必要です。
- 製造プロセスや設備の大きな変更
- 空調システム(HVAC)の改修
- 長期間の製造休止後の再開時
- 環境モニタリングでアラートレベルの逸脱が頻発した場合
定期的な実施により、作業者のスキル維持(適格性維持)を確認するとともに、プロセスの恒常的な安定性を保証します。
まとめ

再生医療における無菌操作は、最新の設備を導入するだけでは達成できません。「人・設備・原材料・方法」の4Mすべてにおいて、リスクベースアプローチに基づいた緻密な管理が必要です。
最終製品の試験には限界があるからこそ、プロセス全体のリスクを可視化し、科学的根拠に基づいた対策(SOP、教育、設備対策)を積み重ねることが、患者様の安全を守る唯一の道です。定期的なリスクレビューとプロセスシミュレーションを通じて、終わりのない品質改善サイクルを回し続けていきましょう。強固な無菌管理体制の構築は、再生医療の未来と信頼を支える土台となるはずです。
無菌操作の基本原則とリスク評価についてよくある質問

無菌操作の基本原則とリスク評価について、現場の実務担当者からよく寄せられる質問とその回答をまとめました。日々の業務や手順書の見直しにお役立てください。
よくある質問
-
Q1. リスク評価はどのくらいの頻度で見直すべきですか?
- 基本的には年1回の定期レビューが推奨されますが、製造工程や設備の変更時、逸脱発生時、法規制の改正時などにはその都度見直しが必要です。リスクマネジメントは一度きりではなく、継続的なプロセスとして運用しましょう。
-
Q2. 無菌操作の教育訓練はどのように行えば効果的ですか?
- 座学だけでなく、実際の現場や模擬環境でのOJTが不可欠です。特に、培地充填試験への参加や、蛍光試薬を用いた発塵・飛散の可視化デモなどは、作業者の意識向上に非常に効果的です。また、定期的なスキル評価を行い、認定制度を設けることも有効です。
-
Q3. グレードAエリアでの「介入操作」とは具体的に何を指しますか?
- 通常の製造フローにはない、修正や調整のための操作を指します。例えば、倒れたバイアルを直す、詰まったチューブを調整する、落下した器具を拾う(拾った後の処理含む)などが該当します。これらは汚染リスクが高いため、可能な限り排除するか、厳格な手順を定める必要があります。
-
Q4. 環境モニタリングで菌が検出された場合、すぐに製造中止すべきですか?
- 検出された菌の種類、数、場所、および傾向(トレンド)によります。アラートレベル(警戒値)であれば調査と監視強化を行いますが、アクションレベル(処置基準)を超えた場合や、無菌性に直結する重大な逸脱の場合は、製造中止や製品の隔離を含めた迅速な対応が必要です。
-
Q5. クローズドシステム(閉鎖系)を導入すれば、グレードA環境は不要ですか?
- 完全に閉鎖されたシステムであっても、接続時などの一部の操作で外部環境に露出するリスクがある場合は、適切な清浄度環境が必要です。ただし、リスク評価に基づき、アイソレーター内やグレードC/D背景での運用が可能になるケースも多く、設備コストと運用リスクのバランスで決定されます。